| | |  |
| | | |
| | | |
Artjom V. Sokirko
The problem of applicability of the mass action law to very small thermodynamic systems originally studied by Blumenfeld et. al. [1] is critically analyzed. The simple kinetic model which results include the results of the statistical mechanics model of [1] as a partial case is proposed. It is shown that in order to obtain the mass action law breakdown being predicted in [1], it is necessary either to consider a non-equilibrium thermodynamic system or to involve a non-thermodynamic mechanism, for example, Maqswell's demon. Conditions, when a statistic function can describe a behaviour of a small thermodynamic system in an adequate way, are discussed. Conclusion about a non-satisfactory explanation of experimental data given in [1] has been made.
Introduction.
Biological cell and organels usually have a size large enough for approximating them thermodynamically instead of using an exact quantum mechanical approach. For example, one of the smallest studied biological objects that can function independently contents at least 10 S UP5(6) ö 10 S UP5(7) molecules of water. It is quite clear that for such a system statistical approach is correct and description by thermodynamic function gives results with an accuracy of 10 S UP5(-3)ÿö 10 S UP5(-4).
On the other hand a real biological system normally consists from a large number of similar objects. It means that even if processes in each system are characterized by discreet probabilities, the average values for the whole system are approximately continuous. For example, during one second a receptor can accept zero or one quant. The average value of quants per one receptor is a certain real value between zero and one. According to the ergodic hypotheses the same results can be obtained after averaging of a single system behaviour over a sufficiently long time period.
At last a statistical mechanics approach based on the concept of the partition function which can be written formally for any system with an arbitrary number of particles is given. After that calculation of all functions, for example, of an average number of particles in a system with chemical equilibrium and of free energy, takes a formal and definite character.
All above mentioned reasons give a good explanation of why the thermodynamic approach is always used for the description of biological systems. For example, if we obtain from calculations that the equilibrium free energy of some substance in the compartment is higher than in the external media, it means that there is a kind of transfer of substance outside depending mainly on permeability of walls [2]. At the same time significant derivations from classic statistical mechanics were observed such as a breakdown of the mass action law [1].
On one hand, the partition function appears in thermodynamic analysis of small systems and is normally simple and compact. On the other hand, a simple operation of averaging which is very clear for big systems, becomes sometimes dangerous and complicated being still simple mathematically. The main question is: "Is the time period of observation (or the number of objects) large enough so that average values of statistic functions show correctly a qualitative behaviour of the system?" Otherwise, if under the period of time given only a few elementary events are possible in the system, it is necessary to analyze each event separately for getting an adequate description of the system.
In the present paper we are going to analyze in details the behaviour of the small system previously described in [1]. First of all we will give a sketch of statistical mechanics calculations of authors [1] which give some paradox results and can not be explained in frames of this approach. Then we will give a simple kinetic model of a little bit more general system which does not lead to any strange results. In the discussion we will show exactly what kind of incorrect assumption leads to the above mentioned conclusions.
The model.
Let us consider the closed volume V separated from the external media by a neutral membrane with a very low permeability. It can be some biological vesicle, for example, tilacoid. We are going to study the recombination-dissociation reaction of water
2 H S DO3(2)0 O() H S DO3(3)0 S UP5(+) + 0H S UP5(-), (1)
inside the volume V. The water product (the product of concentrations of H S DO3(3)0 S UP5(+) and 0H S UP5(-) ions) in the bulk solution is approximately K=10 S UP5(-14). For a neutral solution it gives the concentration of H S DO3(3)0 S UP5(+) or 0H S UP5(-) ions equal to 10 S UP5(-7) mol/litre, which means that only one water molecule from 10 S UP5(7) ones is dissosiated. If the total number of water molecules inside the volume N is less than 10 S UP5(7) it means that less than one couple of H S DO3(3)0 S UP5(+), 0H S UP5(-) exists inside the volume V. Really it means that sometimes there are no ions, sometimes there is one pare, more seldom two or more. We will describe the number of dissosiated water molecules by m - the stohastic function of time. The algebraic difference of the number of hydrogen ions n+ and hidroxil ions n- gives an electric charge q= n+-n- of the volume given in the elementary charge units. While m can only be integer non-negative number, q can be positive, zero or negative depending on the number of H S DO3(3)0 S UP5(+) and 0H S UP5(-) ions present.
These three numbers N, m, q describe completely the thermodynamic system. We can neglect by electric interactions of ions and consider a system consisting of a mixture of three ideal gases. The partition function is expressed simply as
Z = I SU(;; F((V/V0) S UP5(n+);n+!)) F((V/V0) S UP5(n-);n-!) F((K V/V0) S UP5(no);no!) (2)
Here no is the number of nondissosiated water molecules, V0 is so called "quantum volume" and the sum should be taken over all possible states. The numbers n+, n- and no can be expressed in terms of N, m, q:
n+ = m + max (0, q)
n- = m + max (0, -q) (3)
no = N - 2m - |q|
equilibrium constant K must be interpreted now in the frames of microscopic approach as a characteristic of a single molecule of water
K = exp B( F(- DE;kBT))
where DE is the energy difference between dissosiated and non-dissosiated states of a molecule. Taking into account (3) we can simplify (2) as
Z = B( F(KV;V0)) S UP14(N) I SU(;; F(K S UP5( -2m -|q|);n+! n-! no!)) (4)
The only difference between (4) and the analytic expression given by the ref.[1] is that for the derivation of (3) and (4) scheme (1) has been used, while Blumenfeld et al used the questionable scheme H S DO3(2)0 O() H S UP5(+) + 0H S UP5(-). Indeed, we have to note that in the case N>>1 the difference between calculations by these two schemes is not significant enough and makes thus the above given calculations unnecessary. But we would like to discuss more carefully the meaning of the statement "the sum should be taken over all possible states".
Blumenfeld et al give a reasonable on the face of it suggestion that the inert membrane does not allow the transport of any species, which immediately gives N=const, q=const and the sum should be calculated for the only dynamic variable m.: . All the next conclusions of [1] including the one about the mass action law breakdown in small systems are based on this assumption and are formally logical. For example, it was shown that the most significant devivation from the mass action low takes place for systems with q=0.
The main question which we are going to discuss now is the physical interpretation of the system with q=const.
In order to obtain the average value of n+ or n- we have to provide a satisfactory averaging procedure. If only one volume V was monitored we have to take an average over a long period. "Long period" means that the number of dissosiations (and recombinations) should be much more than one. For example, the constant of the water dissosiation at the room temperature is kd =2.10 S UP5(-5) c S UP5(-1). For the closed vesicle with N=106 water molecules it corresponds to the frequency of dissosiation of about kdN ~0.2 c-1. It means that the term "average concentration" for one vesicle of such kind has no meaning for the period of time less than several seconds. After such a long time most real vesicles can not be considered as closed ones and the assumption q=const may be broken. The necessary period of time can be reduced in L times if L identical vesicles (with the same N and q) are considered simultaneously. As it was shown above, fluctuations of N are much lower than N and have no influence on the result. To the contrary, both q and fluctuation of q are of the order of 1. It means that there is no way to produce and keep a large set of identical vesicles with the same q. However, it is possible to produce a set of vesicles with different q. (in the next section this physically realistic model will be discussed in more details.) In order to choose a subset with a certain value of q we have to use some external device which counts the ions inside each vesicle and then pick up some of them with the right value of q. In literature such device is called "Maqswell devil" and can not be created in reality. For completeness we can say that it is possible to construct such a device in a kind of assumption. One can recall the fact that the mobility in the external electric field is proportional to the q number and to use electrofores for separation of vesicles with different q. Of course, such trick is beyond the frames of the equilibrium thermodynamics and is very questionable in sense of an experimental realization, but this is the only way for choosing of identical vesicles we can imagine.
Unfortunately, introducing of a large set of vesicles does not remove the main obstacles created by the assumptions N÷const, q=const, because the time period when there is no ions transfer through membranes of all vesicles decreases proportionally to their number.
The next section is devoted to construction of a simple kinetic model which allows to take into account a slow ions transport through membranes.
Kinetic model.
The main purpose of this section is to construct the simplest kinetic model because we are interested in the general properties of small systems.
Let us assume that the volume V is so small that all water molecules keep a certain order and a quasicristal structure exists everywhere. If we choose the size of water molecules r as the length scale, we have an approximate relation V~Nr3.
Inside the volume V four different events are possible:
1) dissosiation of water molecules;
2) recombination of an existing couple of OH- and H3O+ ions;
3) entering of a proton (possible only when hydrogen ion is situated on the external boundary of the vesicle);
4) exit of a proton (possible only when hydrogen ion is situated near the inner boundary).
There is no specific restriction on the choice of the time scale. For example, it may correspond to the characteristic period of oscillation of water molecules which is of the order of 10-10 c-1. Other possibility is to use as a time scale the average period under which proton joints a certain water molecule. This time is about the characteristic time t of recombination OH- and H3O- ions t =1/krcw ÿ 10-9c, where kr is the constant rate of recombination, cw is the concentration of water. As it has been discussed above, the characteristic period for water dissosiation is 0.1c and for ion transport through a membrane is 1c, so the period t is small enough to assume that only one of events 1)-4) can take place during each period t .
Let us find probabilities of occurrence of different events during the time period t .
The probability of dissosiation of a single water molecule during one second is simply kd, which gives for the volume of N molecules for the period t the probability b:
b = N kdt . (6)
As far as ions H3O+ are mainly reflected from walls, we can assume that ions move inside chaotically. For the occurrence of recombination in a certain point ions H3O+ and OH- must meet each other there, and for the whole vesicle we have
f = F(n+;N) F(n-;N) N = F(n+ n-;N). (7)
The probability g of the ion transfer through the membrane from the external solution into the vesicle is proportional to its concentration cout and the area of a membrane S
g = D cout S (8)
here D is the proportionality coefficient depending on the structure of the membrane, its thickness etc. It can be considered as a kind of diffusion coefficient. Along similar lines we can write down the probability of exit of an ion outside:
h = O(D;Ð) F(n+;N) S (8)
Due to some reasons O(D;Ð) may differ from D. Coefficients O(D;Ð) and D may be interpreted as the renormed diffusion coefficients.
Note that the probabilities of the direct processes - dissosiation and entry - do not depend on the state of the system (i.e. number of ions), while probabilities of the reverse processes - recombination and exit - increase with increasing of those values. It creates a dynamic equilibrium for our kinetic system.
The factors in expressions for f and h that do not depend on number of ions are O(f;^) and O(h;^)
O(h;^) = F( O(D;Ð)S ;N) O(f;^) = F(1;N) (10)
Let us describe the state of the system at a certain moment by the couple of numbers (m,q). Let t is the large enough period of time used for the calculation of the averages; tmq is the time system stays in the state (m,q) and amq º;tmq/t is the probability to find a system in the state (m,q). All values amq satisfy the normalizing condition:
I SU(m=0;N; ) I SU(q=-N;N; amq) = 1. (11)
The probability of transfer in each period t from the state (m,q) to the state (i,j) will be described by the component of tensor P S(ij;mq). As far as only one event can occur at one period of time, the "free walking" process of the system is the Markov's process. It is easy to show that P S(ij;mq) is defined by the set of relations:
P S(m+1 q;m q) = b
P S(m q;m +1q) = (m + |q| + 1)(m + 1) O(f;^)
P S(m q+1;m q) = g q ò 0
P S(m q;m+1 q+1) = g q < 0 (12)
P S(m q;m q+1) = (q+1) O(h;^) q ò 0
P S(m +1q+1;m q) = |q| O(h;^) q < 0
All others P S(ij;mq), except P S(mq;mq)
P S(mq;mq) = I SU((ij)ã(mq);; P S(ij;mq))
- the probability to stay in the same state - are equal to zero. .
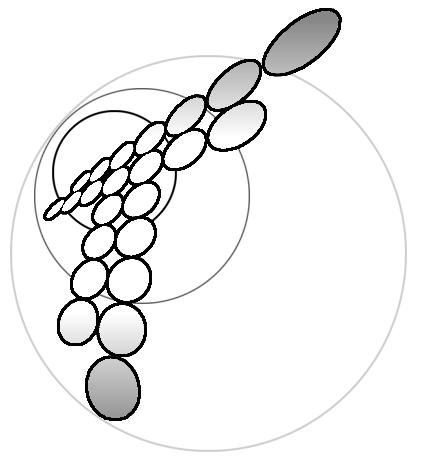
The relation (11) is illustrated by the scheme (Fig.1).
The scheme in the Fig.1 only serves as an illustration to (11) and we present only its most important part because this scheme is infinite upward, backward and downward but not upward. This scheme has no symmetry because it is assumed that only proton transfer can occur whereas OH- transfer can not.
In order to find the unknown value amq we apply the rule of the detailed equilibrium to each state (m,q)
I SU(ij;; aij P S(ij;mq)) = amq ( 1 - P S(mq;mq) ) (13)
This expression has a very simple meaning: left side is the probability to enter the state (m,q) from all other states; ( 1 - P S(mq;mq) ) is the probability to leave this state and the whole right part is the mathematical expectation of leaving. As far as the system is in the steady state, these two numbers must be equal.
Equations (13) for all (m,q) together with normalizing condition (10) give an infinite system for determination of the infinite number of aij. Nevertheless this system can easily be solved with any required accuracy. The reason of it is that the main state of the system is (0,0). The statistical weight of the other states decrease exponentially with the distance from (0,0).
The solution procedure can be defined as follows:
1) choosing a part of the scheme from the Fig.1 and cutting out the rest;
2) solving the system of linear equations;
3) chosing the bigger part of scheme that includes the previous one;
4) solving it;
5) stopping the procedure when the convergence required is achieved.
At the first step we choose the part consisting of 6 cells inside the dash line and the part shown inside the wavy line at the second one. It was shown that even such a small set of states gives a perfect convergence. That is why we are going to present a solution for a six-state system only.
Equations (13) are written as follows:
a10 O(f;^) + a01 h = a00 (b + g)
a00 g + a01 2 O(f;^) = a01 (b + O(h;^) )
a01 b + a10 g = a11 ( 2 O(f;^) + O(h;^) )
a10 O(h;^) + a1-1 2 O(f;^) = a0-1 (b + O(h;^) ) (14)
a0-1 b = a1-1 2 O(f;^)
We have written down only five equations and droped the sixth one, corresponding to the state (0,1), as far as it is just a linear combination of the previous five ones. The normalization condition (10) looks like
a00 + a01 + a10 + a11 + a0-1 + a1-1 =1 (15)
The system of six linear equations (14),(15) can now be solved respectively six unknown variables aij.
The mathematical expectations of the number of OH- and H3O+ ions inside the volume are
M+ = a1-1 +a01 + a10 + 2 a11
M- = a0-1 +a01 + a10 + 2 a1-1 (16)
Discussion.
By choosing some reasonable values of parameters we can estimate the properties of the system. The most interesting for us thing is the concentration of ions inside when its average value (mathematical expectation) is small. In order to find it we have solved the system (14)-(15) for different N and calculated M+/N and M-/N . For the natural case D = O(D;Ð) the results are quite trivial, i.e. concentrations outside and inside are exactly the same. No decreasing of the concentration with the changing of the vesicle size has been observed. The calculations for the bigger system with the higher number of possible states (Fig.1) also gave nothing interesting.
The reason of getting these results is obvious: the volume of the vesicle is in the kinetic (thermodynamic) equilibrium with the external space. The equilibrium situation occurs for all values of the transfer coefficients D = O(D;Ð). If there is any exchange between the inner and the external parts, after sufficiently long time concentrations in both of them will be exactly the same. This trivial result has been proved by solution of the system (14),(15) for arbitrarily small D= O(D;Ð) >0.
The situation is completely different in the case of D = O(D;Ð)=0 exactly. In this case equations (14) look like
a00 = O(f;^) /b a10
a01 = 2 O(f;^) /b a11 (17)
a0-1 = 2 O(f;^) /b a1-1
Two other equations are the consequence of these ones. It means that the system of four equations (17),(15) for six variables aij. requires two additional relations. Actually, putting D = O(D;Ð)=0 separates all columns in the fig.1 from each other. As far as the only process still taking place is dissociation-recombination of water molecules, the charge number becomes permanent for each vesicle. Two additional relations are needed for describing of the initial distributions of vesicles with the charge numbers -1,0 and 1. Of course, here we have some choice. For example, let us say that all vesicles have a zero charge. In this case a01 = a11 = a0-1 = a1-1 = 0 and
M+ = M- = a10 = 2 a11 = (1+ O(f;^) /b ) S UP5(-1) = (1+K cw N-2 ) S UP5(-1) (18)
depends on N significantly if N is small enough: N < R(K cw ). Actually, we arrived to Blumenfeld's results [1] described in the previous section. But the assumption of zero initial charge number is more difficult to fulfil than it seems to be. We have to carry out the preliminary choice of vesicles with q=0, which requires a kind of non-physical device for a large set of vesicles, or choosing of one or a few vesicles and observing them for a time sufficiently short for neglecting proton transfer through the membrane. The last system is described well enough by statistical laws if the number of dissosiations under the observed period is much more than the number of entries/exits. Unfortunately, these events are approximately equally frequent for real objects. Therefore, the area where the results [1] are formally applicable is restricted by vesicles containing about 106 molecules. This value is restricted from above (because the average number of ions inside is less than one) and from below (because the probability of dissosiation is proportional to V which is proportional to N, and the probability of exit/entry is proportional to S which is proportional N2/3). Moreover, the membrane must be thick and hard in order to decrease values D and O(D;Ð). We do not know whether such a system does really exist. The example, discussed in [1], describes exactly this situation, i.e. vesicles spend a long time in the equilibrium with the external solution and there is no preliminary selection done. Protons concentrations inside and outside are equal and the part of vesicles containing a proton can be estimated by the equilibrium equations for a small N
beq ~ F(N; R(K cw )). (19)
Note, that non-equilibrium situation results are significantly different:
bnon ~ F(N;K cw ). (20)
Conclusion.
The present paper was mainly devoted to the discussion of the action mass law breakdown initially claimed in [1,2]. It has been shown that additional strong limitations are required in order to fulfil the necessary conditions of even formal breakdown of the law. Actually, we think that the mass action law is valid with an excellent accuracy for all real system including very small ones. At the same time thermodynamic approach for extremely small systems has very weak limitations. For example, calculations of the average value of the free energy F can be done formally for any system, while the conclusion that a substance transfer should go in the direction of a free energy gradient, strictly speaking, has no background. Fluctuation of F appears to be higher than its average value, which means that the average value of F corresponds to nothing. Therefore, it is better to discuss such small biological systems in the expressive kinetic form without statistic mechanics approach.
Acknowledgments.
Author is thankful to Prof.Yu.A.Chizmadzhev for involving me into this problem and to Dr.A.N.Tikhonov for helpful discussions.
References.
1. Blumenfeld L.A, Grosberg A.Yu and Tikhonov A.N., J.Chem.Phys.,95,7541, 1991.
2.Tikhonov A.N.and Blumenfeld L.A. Zhurnal fisicheskoi khimii, 64, 1729, 1990.
3.Landau L.D., Lifshits E.M., Statistical mechanics.